
Help
- Search
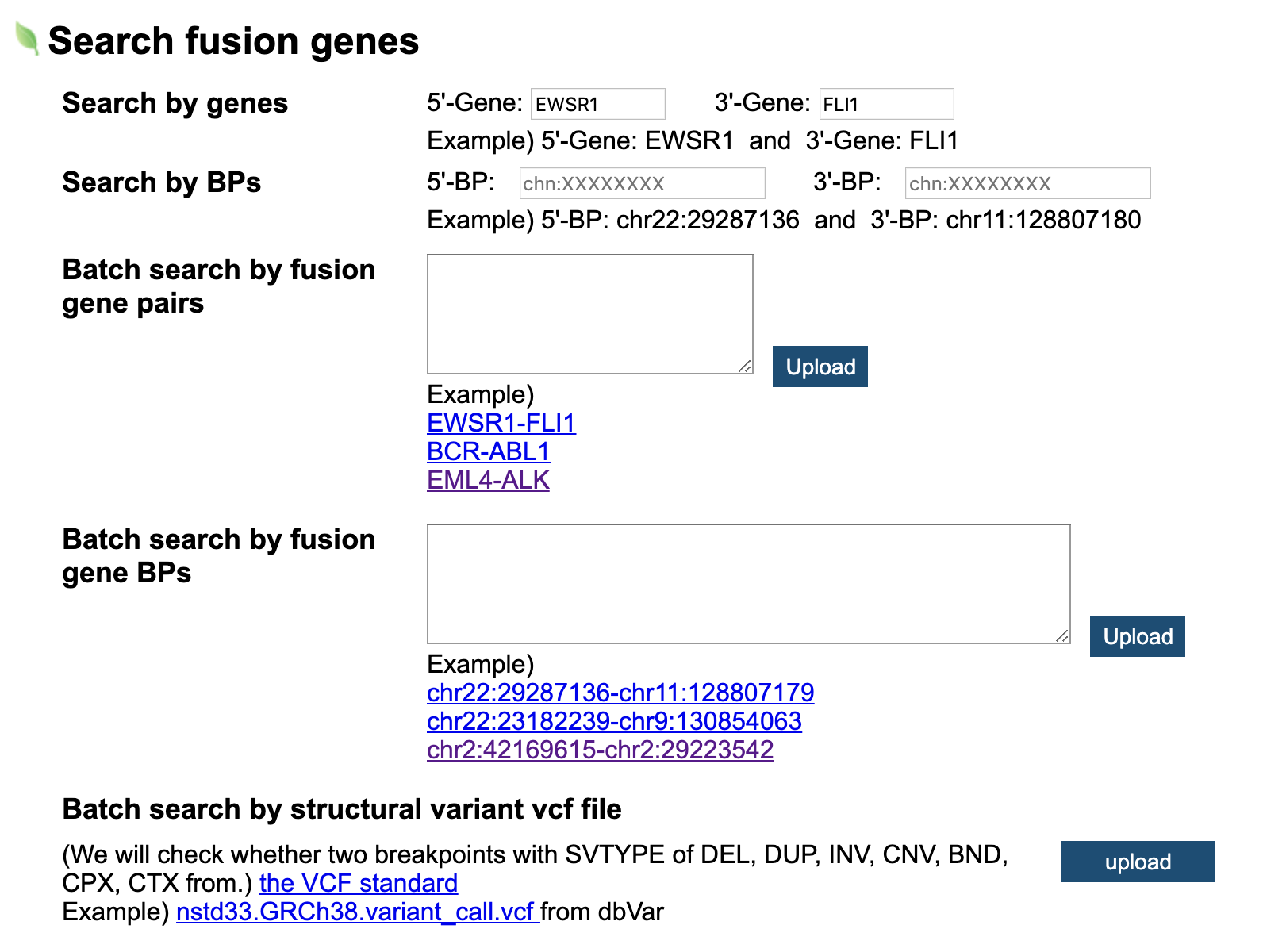
1. The users can use FGviewer by searching individual fusion gene pair symbols or breakpoints (individual search) (a).
2. Users also can use FGviewer through uploading multiple fusion gene pair symbols or multiple breakpoints
(batch search) (b).
3. Users also can use FGviewer through uploading multiple fusion gene pair symbols or multiple breakpoints
(batch search) (b).

- Select the identified fusion genes
1. Clicking the interested fusion gene from this table will show the FGviewer analysis page.
- Browse
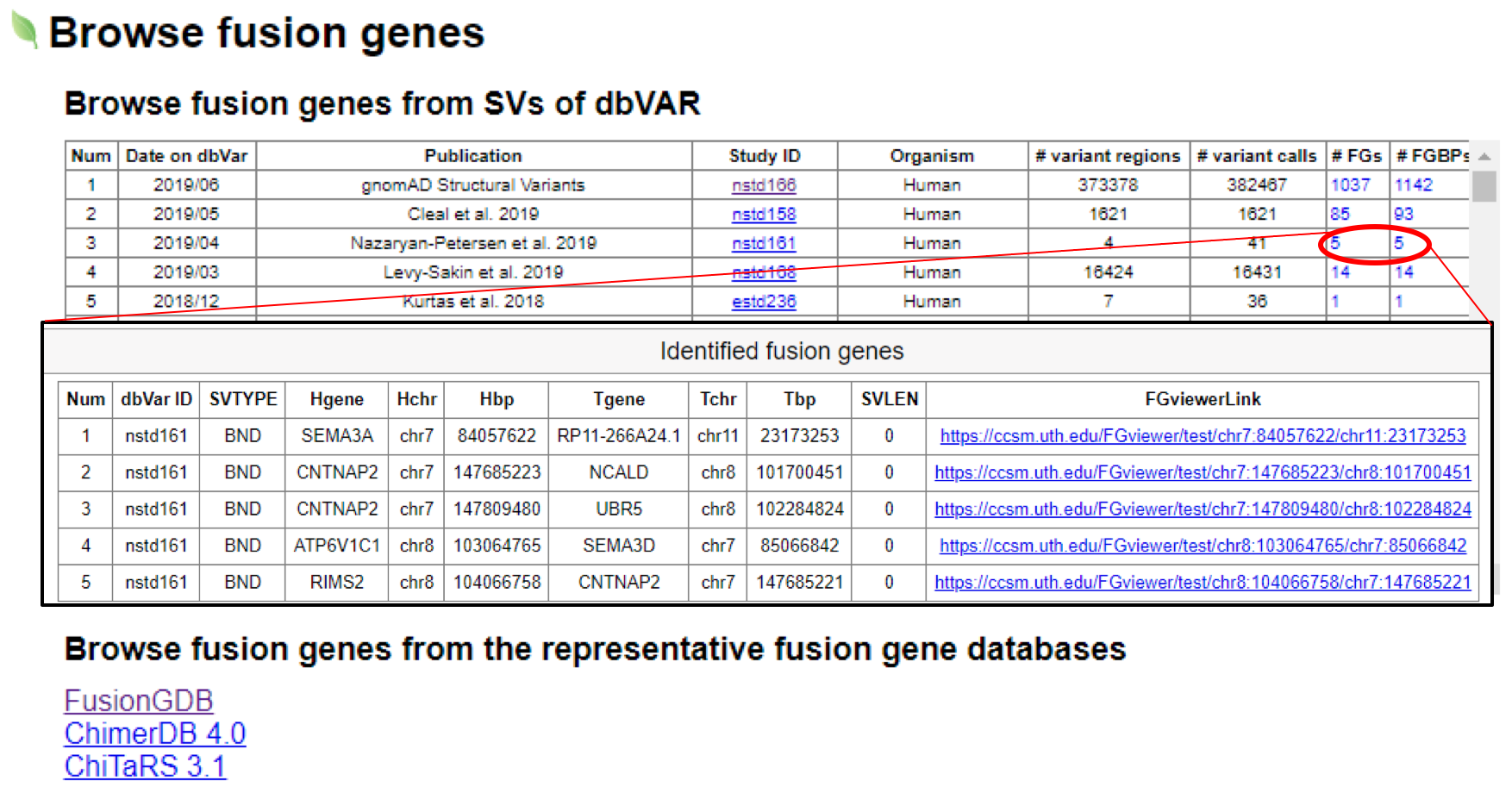
1. dbVar is NCBI’s database of human genomic structural variation including insertinos, deletions, duplications, inversions, mobile elements, translocations, and others. Users can select the interested dbVar study.Then, clicking the number of
fusion
genes or fusion breakpoints will show the detailed fusion gene list with FGviewer hyperlinks in a popup window.
2. There are three representative fusion gene databases including FusionGDB, ChimerDB 4.0, and ChiTaRS. Users can
obtain fusion gene list or fusion gene breakpoint information from these resources, then can use these as the input to
FGviewer to search the potential functional features of those fusion genes.
- FGviewer analysis result
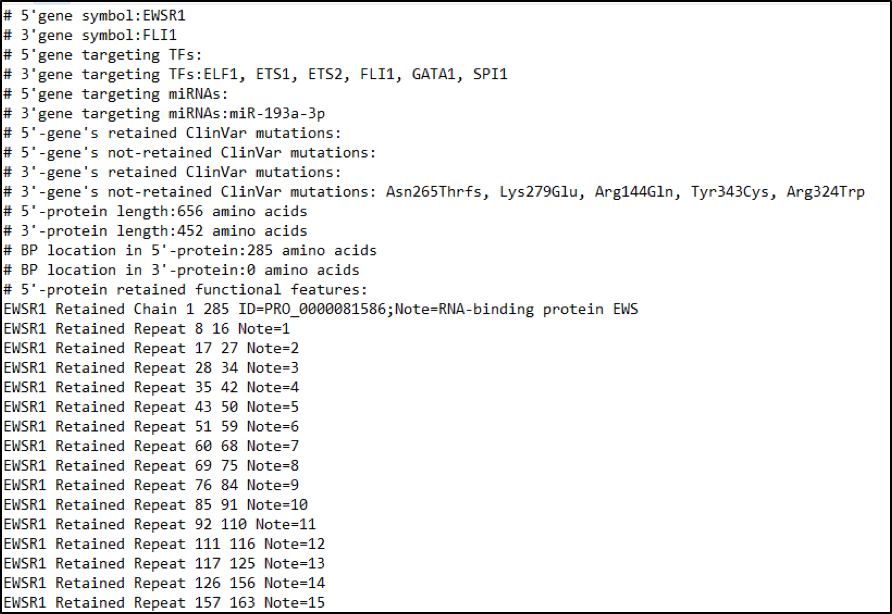
For any combination of gene pairs to be involved in FG, the users can search the functional aspect of FG in
the three bio-molecular levels (DNA-, RNA-, and protein-levels) and one clinical level
(pathogenic-level). The same breakpoint line across four tiers classifies between FG
involving (retained region) and non-involving zone (truncated region) with multiple
types of functional features, such as fusion mRNA and amino acid sequences based
on the user’s breakpoint coordinates, swapped gene expressional regulatory (i.e.,
transcription factor or miRNA binding sites), protein functional features (i.e., protein
domains, protein-protein interactions, binding sites of all molecules, secondary
structure level feature, etc.), clinically relevant variants, etc. To do this, we needed
to arrange genomic-, transcript-, and protein-level coordinates. We hid the intron
structures in the DNA-level and drew the exons only. To set the length of coding
regions same across three levels, the nucleotide length in the DNA- and RNA-levels
were divided by three and made the length of coding regions the same as the length
of the amino acids in the protein level. For the minus stranded genes, we show the
coordinates after performing reverse transcription.

1) When you click the button of 'Save image', then it will provide the option to download png or pdf files as below.

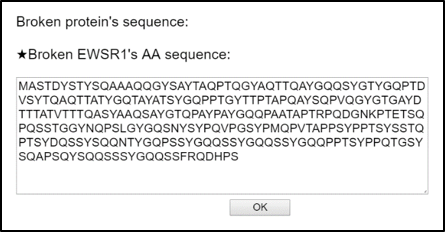
2) When you click the transcript or protein structure bars, then it let you download the broken sequence of nucleotides or amino acids. This sequence is created based on the breakpoint location which is dragged by the user as below.
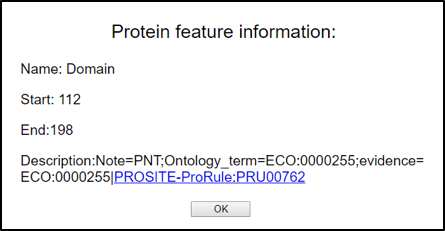
3) When you click the bar of individual functional features, then you can see the detailed information for that feature as below. Protein domain feature provide the link for the PROSITE.
4) When you click the button of 'Download information', then you can download all information of the protein feature retention based on the current breakpoints for both of the genes as below.
2. Used data.
1) Used gene structure model: GENCODE (v28 of GRCh38)
2) Representative protein accessions and sequences: We have downloaded information of 18,963 protein accessions using the query of 'Reviewed human proteins' from UniProt database.
3) Representative transcript accessions and sequences: we have downloaded transcript sequences of 28,860 corresponding transcripts to these protein accessions from Ensembl (GRCh38)
4) Protein feature information for protein-level: We downloaded the protein information in general feature format (GFF) of 18,963 accessions of UniProt database provides 39 protein sequence annotation features based on individual protein sequence including six molecule processing features, 13 region features, four site features, six amino acid modification features, two natural variation features, five experimental info features, and 3 secondary structure features. Detailed information about all of the protein features is in UniProt page . See point (f) of image 3. 1)
5) Transcription factor and target gene pair information for DNA-level: TF-target relationship information were downloaded from TRRUST (version 2), which is a manually curated database of human transcriptional regulatory networks. From TRRUST, we obtained 8427 pairs between 795 TFs and 2492 target genes.
6) microRNA and target gene pair information for mRNA-level: We downloaded the conserved human miRNA-target gene interaction information from TargetScan (release 7.2). For more reliable interactions, we have filtered out the miRNA-target pairs when the context++ score and percent identity were bigger than -0.4 and less than 97%, respectively.
7) Pathogenic variant information of individual genes for pathogenic-level: We downloaded ClinVar data (August 14, 2018) and selected pathogenic variants only.
(UTHealth)
Web File Viewing | Emergency Information |Site Policies